咨詢電話
15618996369
15618996369

做液相色譜的同學基本都會碰到分叉峰,
很多時候,
這都意味著硬件或者方法某些地方可能出現問題。
今天我們就一起看看導致峰分叉的原因,
以及如何解決這個問題。

首先,我們需要判斷,
這到底是一個峰,還是兩個峰?
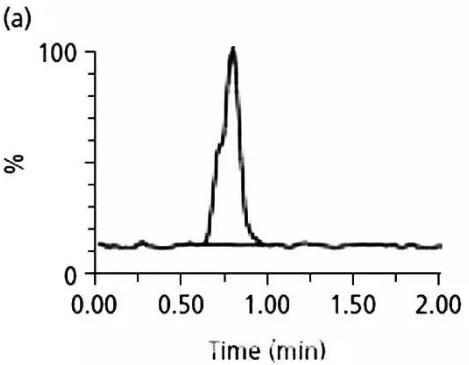
a圖主峰前面有一個肩峰,
這到底是峰形不好,
還是另外一個化合物沒分開呢?
判斷方法
降低該成分在樣品中的濃度
b圖是降低該成分濃度后,
進樣后得到的色譜圖。
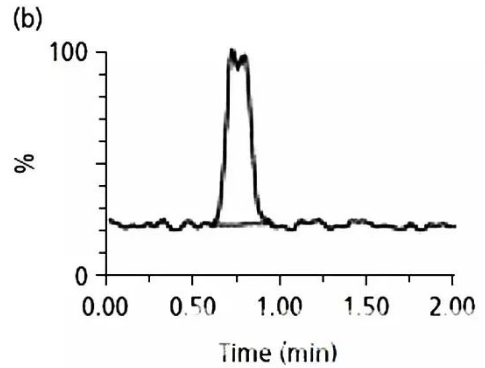
如果這是一個峰,
那肩峰也應該響應變小,
但是這個例子里,
肩峰沒有變小,
這應該是另外一個化合物。
這時候,我們就要考慮如何改變方法
將這兩個東西分開。
如果所有峰的峰形都不好
一般來說,
一個樣品都會出多個峰。
峰形不好,絕大多數時候
在每個峰上面都會出現。
比如像下面這樣:
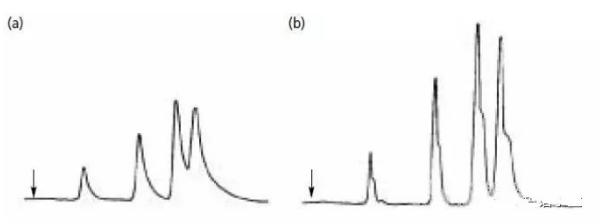
a圖是所有峰都拖尾,
b圖是所有峰都有肩峰,
也可以說峰分叉,
這種情況一般都是色譜柱頭塌陷
或者柱頭的篩板被堵導致的。
為什么會導致峰形問題呢?
請看下面的圖
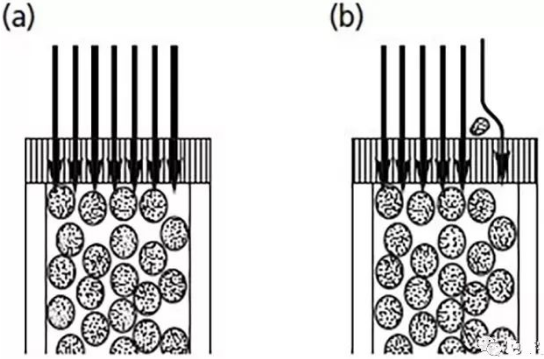
a圖是正常的柱頭,
b圖是篩板被堵的柱頭。
正常情況下,
所有樣品分子都是均勻通過色譜柱,
形成一個對稱的峰形。
堵塞的情況下,
有一部分樣品分子進入色譜柱就會受到阻擾,
導致時間拖后,形成拖尾。
對于色譜柱塌陷的情況,
用同樣的方法大家也不難理解。
只不過這時候,
是一些樣品分子可能跑的比正常的快了一些。
如何解決這個問題呢?
1 反沖
柱出口放空,
沖20-30ml流動相。
雖然柱子上都有表明使用方向,
但是對于硅膠基質的色譜柱
基本都可以反沖。
將柱頭污染物
如泵密封圈的碎屑,
樣品中的污染等沖走,
就能解決問題。
當然,還是要提倡大家注意
樣品上機之前的前處理
和及時更換磨損的泵密封圈。
有條件的還是用保護柱,
大不了就換保護柱,
也比換色譜柱強。
2 更換或清洗篩板
現在估計做這個事情的人不多,
但是誰有廢柱子,
想練練也未嘗不可。
我們一起看看一個實際的例子
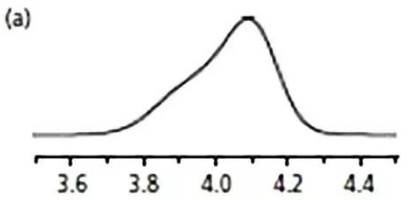
根據之前說的,
可能是柱頭堵了,
或者塌陷了。
但是進一步檢查,
發現另有原因。
方法用的150 mm 4.6 mm, 5 um C18 柱子,
78% 乙腈,1.5ml/min,等度方法,
進樣10ul,100%乙腈溶解的樣品。
一般來說,
雖然10ul的純乙腈不會引起峰形不好,
但是有時候樣品溶劑的極性跟流動相差異較大時,
的確會引起峰形的問題。
我們怎么辦呢?
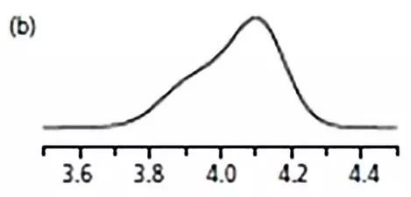
01
從容易的辦法做起,
降低溶劑乙腈的比例,
降到50%看看。
從下圖b,4min的峰可以看出,
沒啥改觀。
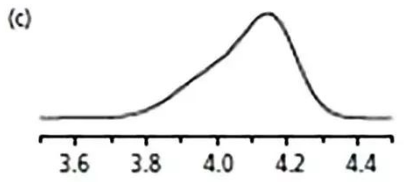
02
接下來反沖柱子,
結果如圖c,
基本沒變化。
算了,明天再說吧。
03
第二天來到實驗室,
還能干嘛呢?
更換篩板?
算了,直接換柱子吧!
結果如d,
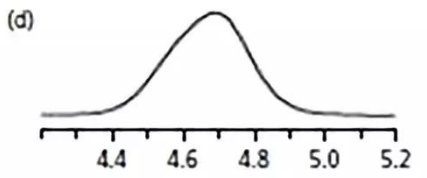
噢,好很多噢。
之前柱子肯定有問題,扔了。
但是峰還是有點寬,
估計還是哪兒有問題。
手上忙,沒時間管這個。
04
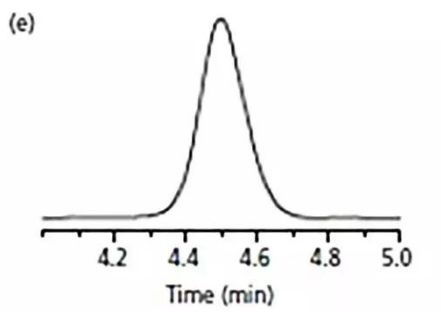
幾天過后,
重新配了流動相,
一跑,好了,如圖e。
 ?
?
雖然到底什么原因導致之前峰形的問題,
已經無從查找
(柱子扔了,舊流動相也沒了),
但是也給我們提供了一個解決問題的步驟:
1
確定峰形問題
是所有峰都有問題,
還是就是某一個峰有問題。
如果所有峰都有問題,
基本都是色譜柱,
或者儀器管路連接的問題。
如果是某一個峰有問題,
那就要從方法上來考慮,
是否需要改變流動相比例,
pH值,色譜柱溫度等等。
2
從jian單的做起
從不需要換什么東西的方法做起。
比如改變樣品溶劑,
反沖色譜柱等等。
3
換東西
換保護柱,換色譜柱,
換流動相(新配),
一步一步的換,
有助于找到問題的根本。
4
總結經驗
問題解決后,
想想需不需要采取什么措施,
防止類似問題的發生,
比如說常換流動相啦,
及時更換密封圈啦,
記錄進樣次數
了解什么時候色譜柱就不行了啊之類的。
希望以上信息能夠讓各位同學
在碰到峰形不好的問題時,
有一些思路,
不至于手足無措。
也歡迎大家留言補充
上面沒有提到的可能影響峰形的可能。
更多檢測標準解讀
專家培訓課程
技術經驗干貨分享
VX關注公眾號:安譜實驗學堂

 微信公眾號
微信公眾號 訪問手機端
訪問手機端